
A protein contact map represents the distance between all possible amino acid residue pairs of a three-dimensional protein structure using a binary two-dimensional matrix. For two residues and , the element of the matrix is 1 if the two residues are closer than a predetermined threshold, and 0 otherwise. Various contact definitions have been proposed: The distance between the Cα-Cα atom with threshold 6-12 Å; distance between Cβ-Cβ atoms with threshold 6-12 Å (Cα is used for Glycine); and distance between the side-chain centers of mass.



Overview
Contact maps provide a more reduced representation of a protein structure than its full 3D atomic coordinates. The advantage is that contact maps are invariant to rotations and translations. They are more easily predicted by machine learning methods. It has also been shown that under certain circumstances (e.g. low content of erroneously predicted contacts) it is possible to reconstruct the 3D coordinates of a protein using its contact map.[1][2]
Contact maps are also used for protein superimposition and to describe similarity between protein structures.[3] They are either predicted from protein sequence or calculated from a given structure.
Contact map prediction
With the availability of high numbers of genomic sequences it becomes feasible to analyze such sequences for coevolving residues. The effectiveness of this approach results from the fact that a mutation in position i of a protein is more likely to be associated with a mutation in position j than with a back-mutation in i if both positions are functionally coupled (e.g. by taking part in an enzymatic domain, or by being adjacent in a folded protein, or even by being adjacent in an oligomer of that protein).[4]
Several statistical methods exist to extract from a multiple sequence alignment such coupled residue pairs: observed versus expected frequencies of residue pairs (OMES);[5] the McLachlan Based Substitution correlation (McBASC);[6] statistical coupling analysis; Mutual Information (MI) based methods;[7] and recently direct coupling analysis (DCA).[8][9]
Machine learning algorithms have been able to enhance MSA analysis methods, especially for non-homologous proteins (ie. shallow MSA's).[10]
Predicted contact maps have been used in the prediction of membrane proteins where helix-helix interactions are targeted.[11]
HB Plot
Knowledge of the relationship between a protein's structure and its dynamic behavior is essential for understanding protein function. The description of a protein three dimensional structure as a network of hydrogen bonding interactions (HB plot)[12] was introduced as a tool for exploring protein structure and function. By analyzing the network of tertiary interactions, the possible spread of information within a protein can be investigated.
HB plot offers a simple way of analyzing protein secondary structure and tertiary structure. Hydrogen bonds stabilizing secondary structural elements (secondary hydrogen bonds) and those formed between distant amino acid residues - defined as tertiary hydrogen bonds - can be easily distinguished in HB plot, thus, amino acid residues involved in stabilizing protein structure and function can be identified.
Features
The plot distinguishes between main chain-main chain, main chain-side chain and side chain-side chain hydrogen bonding interactions. Bifurcated hydrogen bonds and multiple hydrogen bonds between amino acid residues; and intra- and interchain hydrogen bonds are also indicated on the plots. Three classes of hydrogen bondings are distinguished by color-coding; short (distance smaller than 2.5 Å between donor and acceptor), intermediate (between 2.5 Å and 3.2 Å) and long hydrogen bonds (greater than 3.2 Å).
Secondary structure elements in HB plot

In representations of the HB plot, characteristic patterns of secondary structure elements can be recognised easily, as follows:
- Helices can be identified as strips directly adjacent to the diagonal.
- Antiparallel beta sheets appear in HB plot as cross-diagonal.
- Parallel beta sheets appears in the HB plot as parallel to the diagonal.
- Loops appear as breaks in the diagonal between the cross-diagonal beta-sheet motifs.
Examples of usage
Cytochrome P450s
The cytochrome P450s (P450s) are xenobiotic-metabolizing membrane-bound heme-containing enzymes that use molecular oxygen and electrons from NADPH cytochrome P450 reductase to oxidize their substrates. CYP2B4, a member of the cytochrome P450 family is the only protein within this family, whose X-ray structure in both open 11 and closed form 12 is published. The comparison of the open and closed structures of CYP2B4 structures reveals large-scale conformational rearrangement between the two states, with the greatest conformational change around the residues 215-225, which is widely open in ligand-free state and shut after ligand binding; and the region around loop C near the heme.

Examining the HB plot of the closed and open state of CYP2B4 revealed that the rearrangement of tertiary hydrogen bonds was in excellent agreement with the current knowledge of the cytochrome P450 catalytic cycle.
The first step in P450 catalytic cycle is identified as substrate binding. Preliminary binding of a ligand near to the entrance breaks hydrogen bonds S212-E474, S207-H172 in the open form of CYP2B4 and hydrogen bonds E218-A102, Q215-L51 are formed that fix the entrance in the closed form as the HB plot reveals.
The second step is the transfer of the first electron from NADPH via an electron transfer chain. For the electron transfer a conformational change occurs that triggers interaction of the P450 with the NADPH cytochrome P450 reductase. Breaking of hydrogen bonds between S128-N287, S128-T291, L124-N287 and forming S96-R434, A116-R434, R125-I435, D82-R400 at the NADPH cytochrome P450 reductase binding site—as seen in HB plot—transform CYP2B4 to a conformation state, where binding of NADPH cytochrome P450 reductase occurs.
In the third step, oxygen enters CYP2B4 in the closed state - the state where newly formed hydrogen bonds S176-T300, H172-S304, N167-R308 open a tunnel which is exactly the size and shape of an oxygen molecule.
Lipocalin family
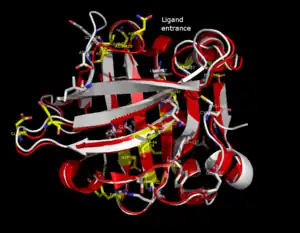
The lipocalin family is a large and diverse family of proteins with functions as small hydrophobic molecule transporters. Beta-lactoglobulin is a typical member of the lipocalin family. Beta-lactoglobulin was found to have a role in the transport of hydrophobic ligands such as retinol or fatty acids.[13] Its crystal structure were determined [e.g. Qin, 1998] with different ligands and in ligand-free form as well. The crystal structures determined so far reveal that the typical lipocalin contains eight-stranded antiparallel-barrel arranged to form a conical central cavity in which the hydrophobic ligand is bound. The structure of beta-lactoglobulin reveals that the barrel-form structure with the central cavity of the protein has an "entrance" surrounded by five beta-loops with centers around 26, 35, 63, 87, and 111, which undergo a conformational change during the ligand binding and close the cavity.
The overall shape of beta-lactoglobulin is characteristic of the lipocalin family. In the absence of alpha-helices, the main diagonal almost disappears and the cross-diagonals representing the beta-sheets dominate the plot. Relatively low number of tertiary hydrogen bonds can be found in the plot, with three high-density regions, one of which is connected to a loop at the residues around 63, a second is connected to the loop around 87, and a third region which is connected to the regions 26 and 35. The fifth loop around 111 is represented only one tertiary hydrogen bond in the HB plot.
In the three-dimensional structure, tertiary hydrogen bonds are formed (1) near to the entrance, directly involved in conformational rearrangement during ligand binding; and (2) at the bottom of the "barrel". HB plots of the open and closed forms of beta-lactoglobulin are very similar, all unique motifs can be recognized in both forms. Difference in HB plots of open and ligand-bound form show few important individual changes in tertiary hydrogen bonding pattern. Especially, the formation of hydrogen bonds between Y20-E157 and S21-H161 in closed form might be crucial in conformational rearrangement. These hydrogen bonds lie at the bottom of the cavity, which suggests that the closure of the entrance of a lipocalin starts when a ligand reached the bottom of the cavity and broke hydrogen bonds R123-Y99, R123-T18, and V41-Q120. Lipocalins are known to have very low sequence similarity with high structural similarity. The only conserved regions are exactly the region around 20 and 160 with an unknown role.
See also
References
- ↑ Pietal, MJ.; Bujnicki, JM.; Kozlowski, LP. (Jun 2015). "GDFuzz3D: a method for protein 3D structure reconstruction from contact maps, based on a non-Euclidean distance function". Bioinformatics. 31 (21): 3499–505. doi:10.1093/bioinformatics/btv390. PMID 26130575.
- ↑ Vassura M, Margara L, Di Lena P, Medri F, Fariselli P, Casadio R (2008). "Reconstruction of 3D Structures From Protein Contact Maps". IEEE/ACM Transactions on Computational Biology and Bioinformatics. 5 (3): 357–367. doi:10.1109/TCBB.2008.27. PMID 18670040. S2CID 6080543.
- ↑ Holm L, Sander C (August 1996). "Mapping the protein universe". Science. 273 (5275): 595–603. Bibcode:1996Sci...273..595H. doi:10.1126/science.273.5275.595. PMID 8662544. S2CID 7509134.
- ↑ Fitch, W. M.; Markowitz, E. (1970). "An improved method for determining codon variability in a gene and its application to the rate of fixation of mutations in evolution". Biochem. Genet. 4 (5): 579–593. doi:10.1007/bf00486096. PMID 5489762. S2CID 26638948.
- ↑ Kass, I.; Horovitz, A. (2002). "Mapping pathways of allosteric communication in GroEL by analysis of correlated mutations". Proteins. 48 (4): 611–617. doi:10.1002/prot.10180. PMID 12211028. S2CID 40289209.
- ↑ Gobel, U.; et al. (1994). "Correlated mutations and residue contacts in proteins". Proteins. 18 (4): 309–317. doi:10.1002/prot.340180402. PMID 8208723. S2CID 14978727.
- ↑ Wollenberg, K. R.; Atchley, W. R. (2000). "Separation of phylogenetic and functional associations in biological sequences by using the parametric bootstrap". Proc. Natl. Acad. Sci. USA. 97 (7): 3288–3291. Bibcode:2000PNAS...97.3288W. doi:10.1073/pnas.97.7.3288. PMC 16231. PMID 10725404.
- ↑ Weigt, M; White, RA; Szurmant, H; Hoch, JA; Hwa, T (2009). "Identification of direct residue contacts in protein–protein interaction by message passing". Proc Natl Acad Sci USA. 106 (1): 67–72. arXiv:0901.1248. Bibcode:2009PNAS..106...67W. doi:10.1073/pnas.0805923106. PMC 2629192. PMID 19116270.
- ↑ Morcos, F; et al. (2011). "Direct-coupling analysis of residue coevolution captures native contacts across many protein families". Proc Natl Acad Sci USA. 108 (49): E1293–E1301. doi:10.1073/pnas.1111471108. PMC 3241805. PMID 22106262.
- ↑ Hanson, Jack; Paliwal, Kuldip K; Litfin, Thomas; Yang, Yuedong; Zhou, Yaoqi (2018). "Accurate Prediction of Protein Contact Maps by Coupling Residual Two-Dimensional Bidirectional Long Short-Term Memory with Convolutional Neural Networks". Bioinformatics. 34 (23): 4039–4045. doi:10.1093/bioinformatics/bty481. PMID 29931279. S2CID 49335891.
- ↑ Lo A, Chiu YY, Rødland EA, Lyu PC, Sung TY, Hsu WL (2009). "Predicting helix-helix interactions from residue contacts in membrane proteins". Bioinformatics. 25 (8): 996–1003. doi:10.1093/bioinformatics/btp114. PMC 2666818. PMID 19244388.
- ↑ Bikadi Z, Demko L, Hazai E (2007). "Functional and structural characterization of a protein based on analysis of its hydrogen bonding network by hydrogen bonding plot". Arch Biochem Biophys. 461 (2): 225–234. doi:10.1016/j.abb.2007.02.020. PMID 17391641.
- ↑ Pérez, M. D.; Calvo, M (1995). "Interaction of beta-lactoglobulin with retinol and fatty acids and its role as a possible biological function for this protein: A review". Journal of Dairy Science. 78 (5): 978–88. doi:10.3168/jds.S0022-0302(95)76713-3. PMID 7622732.
External links
- DISTILL— prediction of protein structural features (including protein residue contact maps)
- Structural Proteomics Tools — includes amino acid contact maps
- ProfCon — prediction of inter-residue contacts
- TMHcon — prediction of helix-helix contacts specifically within the transmembrane parts of membrane proteins
- TMhit — A new transmembrane helix-helix interaction prediction method based on residue contacts
- CMAPpro — A protein contact map prediction server
- —A Tool for Protein Contact-Map Visualization in jerseysforcheapshop