 | |
| Names | |
|---|---|
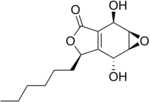
| Preferred IUPAC name
(1aR,2R,5R,6R,6aS)-6-Hexyl-2,6-dihydroxy-2,5,6,6a-tetrahydrooxireno[2,3-f] [2]benzofuran-3(1aH)-one | |
| Other names
(+)-Integrasone | |
| Identifiers | |
| |
3D model (JSmol) |
|
| ChEMBL | |
| ChemSpider | |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C14H20O5 | |
| Molar mass | 268.309 g·mol−1 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |
Integrasone is a polyketide natural product, isolated from an unknown fungus, that has been shown to inhibit the HIV-1 integrase enzyme.[1]
Occurrence
Integrasone occurs naturally in an unidentified sterile fungus. This fungus has been given the label MF6836 by Merck researchers, and was grown on a vermiculite-based solid media, AD2. The methyl ethyl ketone extract of the fungal growth on Sephadex LH 20 (a liquid chromatography medium designed for the separation of small natural products) was then run through both gel permeation chromatography and high performance liquid chromatography to isolate integrasone, which was separated as an amorphous powder at a concentration of 1.8 g/L.[1]
Biological activity
Integrasone inhibits HIV-1 integrase, the viral enzyme responsible for integrating the HIV provirus into the host genome.[2] Integrase accomplishes this using two reactions: 3-prime end processing, and the strand transfer reaction. In the first of these two reactions, the viral DNA is processed by the removal of two deoxynucleotides. In the strand transfer reaction, these processed 3-prime viral DNA ends are covalently bound to the host chromosomal DNA.[3] Integrasone inhibits HIV integrase activity by interfering with the strand transfer reaction, with an IC50 (half maximal inhibitory concentration) of 41 μM.
Due to its high mutation rate and systematic elimination of key immune system cells, HIV is a very difficult virus for which to make a vaccine. In some trials, groups given experimental HIV vaccines have actually had higher incidence of HIV infection than groups given a placebo.[4] As vaccination, the traditional method of fighting viral diseases, is largely unavailable, chemotherapy becomes a better option. Unfortunately, the extraordinarily high mutation rate of HIV allows it to evolve in order to evade both the human immune system and the effect of anti-viral drugs. For this reason, new antiviral HIV-1 drugs are necessary to continue the fight against HIV. The method of inactivation which integrasone uses shows promise for halting the spread of HIV in its host, though it will not eliminate the virus entirely.
Synthesis
The total laboratory synthesis of integrasone has been worked out, starting with a commonly available Diels-Alder adduct of p-benzoquinone and cyclopentadiene. Using a base mediated epoxidation reaction, structure 3 was achieved, which led to structure 4 after exhaustive hydroxymethylation in the presence of DBU. The formation of structure 4 is particularly impressive – it not only forms two important C-C bonds in one step, but also occurs in quantitative yield. Using a retro Diels-Alder reaction, structure 5 was formed in near quantitative yield.[5] Structure 5 was desymmetrized through an enzymatic transesterification process, using an immobilized lipase PS 30 enzyme to give structure 6, which was formed with a 99% enantiomeric excess.[6]

The stereochemistry of the hydroxyl group at carbon 6 in the final integrasone molecule (3) was determined by reduction, which was both regio and stereo selective due to the directing effects of the primary hydroxyl group (carbon 8) and the epoxide ring (carbons 4 and 5). The hydroxy group on carbon 8 is then selectively protected with as the triethylsilyl (TES) ether to give structure 8. With the hydroxyl groups on both carbons 1 and 8 protected, it is then relatively straightforward to stereoselectively reduce the carbonyl group on carbon 3 with sodium borohydride to give the diol depicted in structure 9. Before any oxidation reactions could be used, the two newly formed hydroxyl groups were protected with as acetate esters, forming structure 10. The TES protecting group on the carbon 8 hydroxyl was removed without deprotecting any of the other groups, and then the carbon 8 hydroxyl was oxidized with PCC[7] to give the aldehyde shown in structure 11.

The next step involved the installation of a hexyl chain at the aldehyde carbon (carbon 8). This was accomplished using the Grignard reagent hexylmagnesium bromide, and was highly stereoselective – so much so that the chemists reporting this reaction express their “delight”.[8] It is speculated that this stereoselectivity for product 12 is due to the directing influence of the acetate group attached to carbon 6, which migrates during the reaction to carbon 8. Unfortunately, in 42% of the product, the alkyl chain was not installed, and instead the aldehyde was reduced with an accompanying acetate migration to form a triacetate (structure 14). Efforts to improve this step of the synthesis were made by attempting to vary the temperature and solvent. At high temperatures, more of the triacetate 14 was formed, while at low temperatures, the reaction was sluggish.[8]

Structure 12 is very close to the target molecule, 1 – all that remains is to close the 5-membered ring and form a carbonyl. Base hydrolysis was used to remove the remaining acetate protecting groups, resulting in the tetrol depicted in structure 15. Integrasone (1) is then formed in a single step by oxidation of the primary hydroxyl groups and concerted electron cyclization to form the ring, using sodium chlorite catalyzed by TEMPO and bleach.[9] Structure 16 is a transition state proposed to explain the concerted electrocyclic reaction and the carbonyl formation.[10]

References
- 1 2 Herath, K.; Jayasuriya, H.; Bills, G.; Polishook, J.; Dombrowski, A.; Guan, Z.; Felock, P.; Hazuda, D.; Singh, S. Isolation, Structure, Absolute Stereochemistry and HIV-1 Inhibitory Activity of Integrasone, a Novel Fungal Polyketide. J. Nat. Prod. 2004, 67, 872-874.
- ↑ Craigie, R. HIV Integrase, a Brief Overview from Chemistry to Therapeutics. J. Biol. Chem. 2001, 276, 23213-23216.
- ↑ Hazuda, D.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; Miller, M. Inhibitors of Strand Transfer That Prevent Integration and Inhibit HIV-1 Replication in Cells. Science. 2000, 287, 646-650.
- ↑ Harro, C.; Sun, X.; Stek, J. E.; Leavitt, R. Y.; Mehrotra, D. V.; Wang, F.; Bett, A. J.; Casimiro, D. R.; Shiver, J. W.; DiNubile, M. J.; Quirk, E. Safety and Immunogenicity of the Merck adenovirus serotype 5 (MRKAd5) and MRKAd6 Human Immunodeficiency Virus Type 1 trigene vaccines alone and in combination in healthy adults. Clinical and Vaccine Immunology. 2009, 16, 1285-1292.
- ↑ Mehta, G.; Islam, K. Total Synthesis of the Novel Angiogenesis Inhibitors Epoxyquinols A and B. Tetrahedron Lett. 2003, 44, 3569-3572.
- ↑ Mehta, G.; Islam, K. Enantioselective Total Synthesis of (−)-epoxyquinols A and B. Novel, convenient access to chiral epoxyquinone building blocks through enzymatic desymmetrization. Tetrahedron Lett. 2004, 45, 3611-3615.
- ↑ Rodriguez, A.; Nomen, M.; Spur, B.W.;Godfroid, J.J. Selective oxidation of primary silyl ethers and its application to the synthesis of natural products. Tetrahedron Lett. 1999, 40, 5161-5164.
- 1 2 Metha, G.; Roy, S. Enantioselective Total Synthesis of a Novel Polyketide Natural Product (+)-Integrasone, an HIV-1 Integrase Inhibitor. Chem. Commun. (Cambridge). 2005, 25, 3210-3212.
- ↑ Zhao, M.; Li, J.; Mano, E.; Song, Z,; Tschaen, D.M.; Grabowski, E.J.J.; Reider, P.J. Oxidation of Primary Alcohols to Carboxylic Acids with Sodium Chlorite Catalyzed by TEMPO and Bleach. J. Org. Chem. 1999, 64, 2564-2566.
- ↑ Mehta, G; Roy, S. Enantioselective Total Synthesis of (+)-Eupenoxide and (+)-Phomoxide: Revision of Structures and Assignment of Absolute Configuration. Org. Lett. 2004, 6, 2389-2392.