| BENTA Disease | |
|---|---|
| Other names | B-cell expansion with NF-kB and T-cell anergy disease |
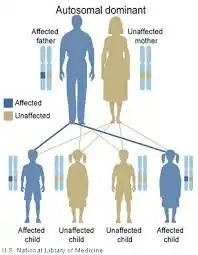 | |
| For each child an affected parent has, there is a 50% chance of passing on the mutation, regardless of the sex of the child autosomal dominant | |
| Specialty | Immunology |
BENTA disease is a rare genetic disorder of the immune system. BENTA stands for "B cell expansion with NF-κB and T cell anergy" and is caused by germline heterozygous gain-of-function mutations in the gene CARD11 (see OMIM entry #607210). This disorder is characterized by polyclonal B cell lymphocytosis with onset in infancy, splenomegaly, lymphadenopathy, mild immunodeficiency, and increased risk of lymphoma. Investigators Andrew L. Snow and Michael J. Lenardo at the National Institute of Allergy and Infectious Diseases at the U.S. National Institutes of Health first characterized BENTA disease in 2012. Dr. Snow's current laboratory at the Uniformed Services University of the Health Sciences is now actively studying this disorder.[1][2]
Presentation
Individuals with BENTA disease have polyclonal B cell lymphocytosis (i.e. excess B cells) developing in infancy, in addition to splenomegaly and lymphadenopathy. Patients may have low serum IgM and mildly anergic T cells. These features likely contribute to the mild immunodeficiency seen with BENTA disease. Patients are generally susceptible to recurrent sinopulmonary and ear infections in childhood, and may be more susceptible to certain viruses including Epstein–Barr virus, BK virus, and molluscum contagiosum.[1]
Genetics
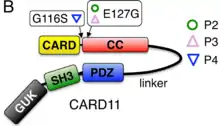
BENTA disease is caused by germline-encoded gain-of-function mutations in the gene CARD11. This is a 138 kB gene mapping to chromosome 7p22 with 26 exons encoding a 1,154 amino acid protein.[3][4] The CARD11 protein (also known as CARMA1) is a scaffolding protein required for NF-κB activation in both B and T lymphocytes. Gain-of-function mutations drive constitutive NF-κB activation in both types of cells. Most mutations are localized within or just upstream of the coiled-coil domain (exons 4–9) of the protein. Patient phenotypes also suggest that B cell differentiation might be partially impaired in BENTA disease, contributing to a low percentage of class-switched and memory B cells.[1][2]
Germline gain-of-function mutations in CARD11 manifest a less severe illness than loss-of-function mutations seen in CARD11 deficiency (OMIM #615206), an autosomal recessive condition manifesting in severe combined immunodeficiency.[1]
The gain-of-function CARD11 mutations associated with BENTA disease may also predispose patients to B cell malignancy. Importantly, overactive NF-κB is frequently associated with B cell malignancy and, specifically, somatic gain-of-function CARD11 mutations are seen frequently in diffuse large B cell lymphoma (DLBCL). However, most BENTA patients present with polyclonal B cell accumulation with no evidence of oligoclonal or monoclonal populations (i.e. malignancy). These mutations do not appear to be associated with T cell malignancies.[2]
Inheritance
This disorder is inherited in an autosomal dominant manner. Autosomal refers to the fact that every person has two CARD11 alleles, one inherited from each parent. This is in contrast to sex-linked chromosomes. Dominant means that the abnormal allele dominates the matching, normal allele. Only one of the two copies (alleles) of CARD11 needs to be abnormal for a person to have BENTA disease. BENTA disease can also arise spontaneously in a patient as the result of a de novo mutations in CARD11, which means that the mutation was not inherited from the parents. In this case the patient could still pass on the mutation to his/her children.
Children of a parent who carries a CARD11 mutation have a 50% chance of inheriting the mutation. In a family, each child's risk of inheriting the mutated CARD11 allele is independent of whether or not other siblings inherited the mutation. For example, if the first four children in a family have the mutation, the next child has the same 50% risk of inheriting the mutation. Children who do not inherit the mutation will not develop BENTA disease nor pass it on to their children.
Diagnosis
The majority of patient peripheral blood mononucleated cells are polyclonal naïve mature B cells, with a significant increase in immature, transitional B cell numbers (identified as CD10+).[5] Percentages of circulating class-switched and memory B cells are very low, and in vitro studies show poor B cell differentiation and immunoglobulin secretion. Serum IgM is low in most patients, while total IgG and IgA may be on the low end of normal. Patients demonstrate defective antibody production against T cell-independent, polysaccharide-based vaccines. Some patients may not mount protective antibody titers to other vaccines, such as measles and varicella zoster virus.[2][6]
T cell counts are generally within or just above the normal range. In vitro stimulation of T cells demonstrates that both CD4+ and CD8+ T cells are less responsive than normal, suggesting mild T cell anergy in patients.[1]
A diagnosis of leukemia can generally be ruled out in these patients based on the unremarkable appearance of small resting lymphocytes in the blood; however, patients must be closely monitored for any signs of monoclonal or oligoclonal B cell expansion because there may be an increased risk for B cell malignancy. Specifically, one patient with BENTA disease was reported as having developed B cell chronic lymphocytic leukemia (B-CLL) as an adult.[1]
Treatment
There is currently minimal therapeutic intervention available for BENTA disease. Patients are closely monitored for infections and for signs of monoclonal or oligoclonal B cell expansion that could indicate B cell malignancy. Splenectomy is unlikely to reduce B cell burden; peripheral blood B cell counts rose significantly in three patients who underwent the procedure. It remains to be determined whether immunosuppressive drugs, including B cell-depleting drugs such as rituximab, could be effective for treating BENTA disease.[1]
References
- 1 2 3 4 5 6 7 Turvey, SE; Durandy, A; Fischer, A; Fung, SY; Geha, RS; Gewies, A; Giese, T; Greil, J; Keller, B; McKinnon, ML; Neven, B; Rozmus, J; Ruland, J; Snow, AL; Stepensky, P; Warnatz, K (2014). "The CARD11-BCL10-MALT1 (CBM) signalosome complex: Stepping into the limelight of human primary immunodeficiency". The Journal of Allergy and Clinical Immunology. 134 (2): 276–84. doi:10.1016/j.jaci.2014.06.015. PMC 4167767. PMID 25087226.
- 1 2 3 4 5 Snow, A. L.; Xiao, W.; Stinson, J. R.; Lu, W.; Chaigne-Delalande, B.; Zheng, L.; Pittaluga, S.; Matthews, H. F.; Schmitz, R.; Jhavar, S.; Kuchen, S.; Kardava, L.; Wang, W.; Lamborn, I. T.; Jing, H.; Raffeld, M.; Moir, S.; Fleisher, T. A.; Staudt, L. M.; Su, H. C.; Lenardo, M. J. (5 November 2012). "Congenital B cell lymphocytosis explained by novel germline CARD11 mutations". Journal of Experimental Medicine. 209 (12): 2247–2261. doi:10.1084/jem.20120831. PMC 3501355. PMID 23129749.
- ↑ "CARD11 caspase recruitment domain family, member 11 [ Homo sapiens (human) ]". NCBI > Genes & Expression > Gene. NCBI. Retrieved 4 September 2014.
- ↑ "Caspase recruitment domain-containing protein 11 [Homo sapiens]". NCBI. Retrieved 4 September 2014.
- ↑ Chung JB, Silverman M, Monroe JG (June 2003). "Transitional B cells: step by step towards immune competence". Trends Immunol. 24 (6): 343–9. doi:10.1016/s1471-4906(03)00119-4. PMID 12810111.
- ↑ Kniffin, Cassandra. "#606445 Persistent polyclonal B-cell lymphocytosis; PPBL". OMIM. Johns Hopkins University. Archived from the original on 24 September 2015. Retrieved 4 September 2014.