H3K27me3 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the tri-methylation of lysine 27 on histone H3 protein.
This tri-methylation is associated with the downregulation of nearby genes via the formation of heterochromatic regions.[1]
Nomenclature
H3K27me3 indicates trimethylation of lysine 27 on histone H3 protein subunit:
| Abbr. | Meaning |
| H3 | H3 family of histones |
| K | standard abbreviation for lysine |
| 27 | position of amino acid residue
(counting from N-terminus) |
| me | methyl group |
| 3 | number of methyl groups added |
Lysine methylation
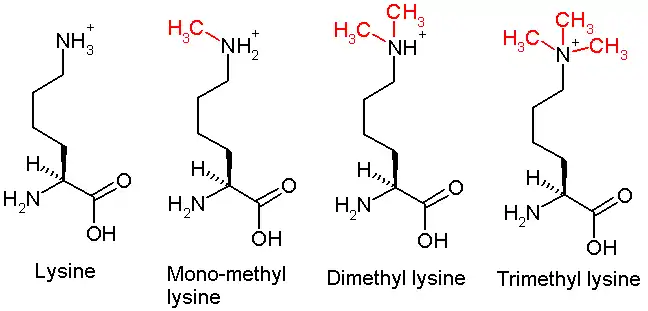
This diagram shows the progressive methylation of a lysine residue. The tri-methylation denotes the methylation present in H3K27me3.
Understanding histone modifications
The genomic DNA of eukaryotic cells is wrapped around special protein molecules known as histones. The complexes formed by the looping of the DNA are known as chromatin. The basic structural unit of chromatin is the nucleosome: this consists of the core octamer of histones (H2A, H2B, H3 and H4) as well as a linker histone and about 180 base pairs of DNA. These core histones are rich in lysine and arginine residues. The carboxyl (C) terminal end of these histones contribute to histone-histone interactions, as well as histone-DNA interactions. The amino (N) terminal charged tails are the site of the post-translational modifications, such as the one seen in H3K27me3.[2][3]
Mechanism and function of modification
The placement of a repressive mark on lysine 27 requires the recruitment of chromatin regulators by transcription factors. These modifiers are either histone modification complexes which covalently modify the histones to move around the nucleosomes and open the chromatin, or chromatin remodelling complexes which involve movement of the nucleosomes without directly modifying them.[4] These histone marks can serve as docking sites of other co-activators as seen with H3K27me3. This occurs through polycomb mediated gene silencing via histone methylation and chromodomain interactions. A polycomb repressive complex (PRC); PRC2, mediates the tri-methylation of histone 3 on lysine 27 through histone methyl transferase activity.[5] This mark can recruit PRC1 which will bind and contribute to the compaction of the chromatin.[6]
H3K27me3 is linked to the repair of DNA damages, particularly repair of double-strand breaks by homologous recombinational repair.[7]
Relationship with other modifications
H3K27 can undergo a variety of other modifications. It can exist in mono- as well as di-methylated states. The roles of these respective modifications are not as well characterised as tri-methylation. PRC2 is however believed to be implicated in all the different methylations associated with H3K27me.
H3K27me1 is linked to promotion of transcription and is seen to accumulate in transcribed genes. Histone-histone interactions play a role in this process. Regulation occurs via Setd2-dependent H3K36me3 deposition.[8]
H3K27me2 is broadly distributed within the core histone H3 and is believed to play a protective role by inhibiting non-cell-type specific enhancers. Ultimately, this leads to the inactivation of transcription.[9]
Acetylation is usually linked to the upregulation of genes. This is the case in H3K27ac which is an active enhancer mark. It is found in distal and proximal regions of genes. It is enriched in Transcriptional start sites (TSS). H3K27ac shares a location with H3K27me3 and they interact in an antagonistic manner.
H3K27me3 is often seen to interact with H3K4me3 in bivalent domains .[10] These domains are usually found in embryonic stem cells and are pivotal for proper cell differentiation. H3K27me3 and H3K4me3 determine whether a cell will remain unspecified or will eventually differentiate.[11][12] The Grb10 gene in mice makes use of these bivalent domains. Grb10 displays imprinted gene expression. Genes are expressed from one parental allele while simultaneously being silenced in the other parental allele.[13]
Other well characterised modifications are H3K9me3 as well as H4K20me3 which—just like H3K27me3—are linked to transcriptional repression via formation of heterochromatic regions. Mono-methylations of H3K27, H3K9, and H4K20 are all associated with gene activation.[14]
Epigenetic implications
The post-translational modification of histone tails by either histone modifying complexes or chromatin remodelling complexes are interpreted by the cell and lead to complex, combinatorial transcriptional output. It is thought that a Histone code dictates the expression of genes by a complex interaction between the histones in a particular region.[15] The current understanding and interpretation of histones comes from two large scale projects: ENCODE and the Epigenomic roadmap.[16] The purpose of the epigenomic study was to investigate epigenetic changes across the entire genome. This led to chromatin states which define genomic regions by grouping the interactions of different proteins and/or histone modifications together. Chromatin states were investigated in Drosophila cells by looking at the binding location of proteins in the genome. Use of ChIP-sequencing revealed regions in the genome characterised by different banding.[17] Different developmental stages were profiled in Drosophila as well, an emphasis was placed on histone modification relevance.[18] A look in to the data obtained led to the definition of chromatin states based on histone modifications.[19] Certain modifications were mapped and enrichment was seen to localize in certain genomic regions. Five core histone modifications were found with each respective one being linked to various cell functions.
- H3K4me3-promoters
- H3K4me1- primed enhancers
- H3K36me3-gene bodies
- H3K27me3-polycomb repression
- H3K9me3-heterochromatin
The human genome was annotated with chromatin states. These annotated states can be used as new ways to annotate a genome independently of the underlying genome sequence. This independence from the DNA sequence enforces the epigenetic nature of histone modifications. Chromatin states are also useful in identifying regulatory elements that have no defined sequence, such as enhancers. This additional level of annotation allows for a deeper understanding of cell specific gene regulation.[20]
Cause-and-effect relationship between sperm-transmitted histone marks and gene expression and development is in offspring and grandoffspring.[21]
Clinical significance
H3K27me3 is believed to be implicated in some diseases due to its regulation as a repressive mark.
Cohen-Gibson syndrome
Cohen-Gibson syndrome is a disorder linked to overgrowth and is characterised by dysmorphic facial features and variable intellectual disability. In some cases, a de novo missense mutation in EED was associated with decreased levels of H3K27me3 in comparison to wild type. This decrease was linked to loss of PRC2 activity.[22]
Diffuse midline Glioma

Diffuse midline glioma, H3K27me3-altered (DMG), also known as diffuse intrinsic pontine glioma (DIPG) is a type of highly aggressive brain tumor mostly found in children. All DMGs exhibit loss of H3K27me3, in about 80 % of cases due to a genetic mutation receplacing lysine with methionine (M), known as H3K27M. In rare forms, H3Kme3-loss is mediated by overexpression of the EZH inhibiting protein, decreasing PRC2-activity.[23]
Spectrum disorders
There is evidence that implicates the downregulation of expression of H3K27me3 in conjunction with differential expression of H3K4me3 AND DNA methylation may play a factor in Fetal Alcohol Spectrum Disorder (FASD) in C57BL/6J mice. This histone code is believed to affect the peroxisome associated pathway and induce the loss of the peroxisomes to ameliorate oxidative stress.[24]
Methods
The histone mark H3K27me3 can be detected in a variety of ways:
1. Chromatin Immunoprecipitation Sequencing (ChIP-sequencing) measures the amount of DNA enrichment once bound to a targeted protein and immunoprecipitated. It results in good optimization and is used in vivo to reveal DNA-protein binding occurring in cells. ChIP-Seq can be used to identify and quantify various DNA fragments for different histone modifications along a genomic region.[25]
2. Micrococcal Nuclease sequencing (MNase-seq) is used to investigate regions that are bound by well positioned nucleosomes. Use of the micrococcal nuclease enzyme is employed to identify nucleosome positioning. Well positioned nucleosomes are seen to have enrichment of sequences.[26]
3. Assay for transposase accessible chromatin sequencing (ATAC-seq) is used to look in to regions that are nucleosome free (open chromatin). It uses hyperactive Tn5 transposon to highlight nucleosome localisation.[27][28][29]
See also
- Histone methylation
- Histone methyltransferase
- Methyllysine
- JARID1B, an enzyme which can reverse the methylation
- Bivalent chromatin, where this repressing modification is often used with activator H3K4me3
References
- ↑ Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stützer A, Fischle W, Bonaldi T, Pasini D (January 2014). "Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity". Molecular Cell. 53 (1): 49–62. doi:10.1016/j.molcel.2013.10.030. hdl:11858/00-001M-0000-0015-367D-4. PMID 24289921.
- ↑ Ruthenburg AJ, Li H, Patel DJ, Allis CD (December 2007). "Multivalent engagement of chromatin modifications by linked binding modules". Nature Reviews Molecular Cell Biology. 8 (12): 983–94. doi:10.1038/nrm2298. PMC 4690530. PMID 18037899.
- ↑ Kouzarides T (February 2007). "Chromatin modifications and their function". Cell. 128 (4): 693–705. doi:10.1016/j.cell.2007.02.005. PMID 17320507.
- ↑ Strahl BD, Allis CD (January 2000). "The language of covalent histone modifications". Nature. 403 (6765): 41–5. Bibcode:2000Natur.403...41S. doi:10.1038/47412. PMID 10638745. S2CID 4418993.
- ↑ Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE (October 2008). "Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains". PLOS Genetics. 4 (10): e1000242. doi:10.1371/journal.pgen.1000242. PMC 2567431. PMID 18974828.
- ↑ Sanz LA, Chamberlain S, Sabourin JC, Henckel A, Magnuson T, Hugnot JP, Feil R, Arnaud P (October 2008). "A mono-allelic bivalent chromatin domain controls tissue-specific imprinting at Grb10". The EMBO Journal. 27 (19): 2523–32. doi:10.1038/emboj.2008.142. PMC 2567399. PMID 18650936.
- ↑ Wei S, Li C, Yin Z, Wen J, Meng H, Xue L, Wang J (2018). "Histone methylation in DNA repair and clinical practice: new findings during the past 5-years". J Cancer. 9 (12): 2072–2081. doi:10.7150/jca.23427. PMC 6010677. PMID 29937925.
- ↑ Edmunds JW, Mahadevan LC, Clayton AL (January 2008). "Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation". The EMBO Journal. 27 (2): 406–20. doi:10.1038/sj.emboj.7601967. PMC 2168397. PMID 18157086.
- ↑ Jones, Peter A.; Archer, Trevor K.; Baylin, Stephen B.; Beck, Stephan; Berger, Shelley; Bernstein, Bradley E.; Carpten, John D.; Clark, Susan J.; Costello, Joseph F.; Doerge, Rebecca W.; Esteller, Manel; Feinberg, Andrew P.; Gingeras, Thomas R.; Greally, John M.; Henikoff, Steven; Herman, James G.; Jackson-Grusby, Laurie; Jenuwein, Thomas; Jirtle, Randy L.; Kim, Young-Joon; Laird, Peter W.; Lim, Bing; Martienssen, Robert; Polyak, Kornelia; Stunnenberg, Henk; Tlsty, Thea Dorothy; Tycko, Benjamin; Ushijima, Toshikazu; Zhu, Jingde; et al. (August 2008). "Moving AHEAD with an international human epigenome project". Nature. 454 (7205): 711–5. Bibcode:2008Natur.454..711J. doi:10.1038/454711a. PMC 6528477. PMID 18685699.
- ↑ Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES (August 2008). "Genome-scale DNA methylation maps of pluripotent and differentiated cells". Nature. 454 (7205): 766–70. Bibcode:2008Natur.454..766M. doi:10.1038/nature07107. PMC 2896277. PMID 18600261.
- ↑ Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES (April 2006). "A bivalent chromatin structure marks key developmental genes in embryonic stem cells". Cell. 125 (2): 315–26. doi:10.1016/j.cell.2006.02.041. PMID 16630819.
- ↑ Huang J, Fan T, Yan Q, Zhu H, Fox S, Issaq HJ, Best L, Gangi L, Munroe D, Muegge K (2004). "Lsh, an epigenetic guardian of repetitive elements". Nucleic Acids Research. 32 (17): 5019–28. doi:10.1093/nar/gkh821. PMC 521642. PMID 15448183.
- ↑ Blagitko N, Mergenthaler S, Schulz U, Wollmann HA, Craigen W, Eggermann T, Ropers HH, Kalscheuer VM (July 2000). "Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue- and isoform-specific fashion". Human Molecular Genetics. 9 (11): 1587–95. doi:10.1093/hmg/9.11.1587. PMID 10861285.
- ↑ Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (May 2007). "High-resolution profiling of histone methylations in the human genome". Cell. 129 (4): 823–37. doi:10.1016/j.cell.2007.05.009. PMID 17512414.
- ↑ Jenuwein T, Allis CD (August 2001). "Translating the histone code". Science. 293 (5532): 1074–80. doi:10.1126/science.1063127. PMID 11498575. S2CID 1883924.
- ↑ Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. (The ENCODE Project Consortium) (June 2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project". Nature. 447 (7146): 799–816. Bibcode:2007Natur.447..799B. doi:10.1038/nature05874. PMC 2212820. PMID 17571346.
- ↑ Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B (October 2010). "Systematic protein location mapping reveals five principal chromatin types in Drosophila cells". Cell. 143 (2): 212–24. doi:10.1016/j.cell.2010.09.009. PMC 3119929. PMID 20888037.
- ↑ Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, et al. (modENCODE Consortium) (December 2010). "Identification of functional elements and regulatory circuits by Drosophila modENCODE". Science. 330 (6012): 1787–97. Bibcode:2010Sci...330.1787R. doi:10.1126/science.1198374. PMC 3192495. PMID 21177974.
- ↑ Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, et al. (March 2011). "Comprehensive analysis of the chromatin landscape in Drosophila melanogaster". Nature. 471 (7339): 480–5. Bibcode:2011Natur.471..480K. doi:10.1038/nature09725. PMC 3109908. PMID 21179089.
- ↑ Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, et al. (Roadmap Epigenomics Consortium) (February 2015). "Integrative analysis of 111 reference human epigenomes". Nature. 518 (7539): 317–30. Bibcode:2015Natur.518..317.. doi:10.1038/nature14248. PMC 4530010. PMID 25693563.
- ↑ University of California, Santa Cruz (27 September 2022). "New study shows transmission of epigenetic memory across multiple generations". Proceedings of the National Academy of Sciences of the United States of America. Phys.org. 119 (40): e2209471119. doi:10.1073/pnas.2209471119. PMC 9546627. PMID 36161922. Retrieved 28 September 2022.
- ↑ Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, et al. (June 2017). "Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome". Human Mutation. 38 (6): 637–648. doi:10.1002/humu.23200. PMID 28229514.
- ↑ Central Nervous System Tumours. International Agency for Research on Cancer. 2022. pp. 69–73. ISBN 9789283245087.
- ↑ Chater-Diehl EJ, Laufer BI, Castellani CA, Alberry BL, Singh SM (2 May 2016). "Alteration of Gene Expression, DNA Methylation, and Histone Methylation in Free Radical Scavenging Networks in Adult Mouse Hippocampus following Fetal Alcohol Exposure". PLOS ONE. 11 (5): e0154836. Bibcode:2016PLoSO..1154836C. doi:10.1371/journal.pone.0154836. PMC 4852908. PMID 27136348.
- ↑ "Whole-Genome Chromatin IP Sequencing (ChIP-Seq)" (PDF). Illumina. Retrieved 23 October 2019.
- ↑ "MAINE-Seq/Mnase-Seq". illumina. Retrieved 23 October 2019.
- ↑ Buenrostro, Jason D.; Wu, Beijing; Chang, Howard Y.; Greenleaf, William J. (2015). "ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide". Current Protocols in Molecular Biology. 109: 21.29.1–21.29.9. doi:10.1002/0471142727.mb2129s109. PMC 4374986. PMID 25559105.
- ↑ Schep, Alicia N.; Buenrostro, Jason D.; Denny, Sarah K.; Schwartz, Katja; Sherlock, Gavin; Greenleaf, William J. (2015). "Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions". Genome Research. 25 (11): 1757–1770. doi:10.1101/gr.192294.115. ISSN 1088-9051. PMC 4617971. PMID 26314830.
- ↑ Song, L.; Crawford, G. E. (2010). "DNase-seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells". Cold Spring Harbor Protocols. 2010 (2): pdb.prot5384. doi:10.1101/pdb.prot5384. ISSN 1559-6095. PMC 3627383. PMID 20150147.